1.0 Introduction.
Hepatitis B virus (HBV) belongs to the Hepadnavirida family, Humans are the only known natural host. HBV enters the liver via the bloodstream, and replication occurs only in liver tissue [1]. About two billion people are infected worldwide with approximately 350 million others suffering the chronic form of the disease [2]. In Africa, more than 50 million people are chronically infected, with mortality risk of about 25% [3]. Infection with HBV leads to a wide spectrum of clinical presentations ranging from an asymptomatic carrier state to self-limited acute or fulminant hepatitis to chronic hepatitis with progression to cirrhosis and hepatocellular carcinoma [4].
Evidence has been accumulating that certain HBV mutants are associated with unique clinical manifestations, may affect the natural course of the infection and confer resistance to antiviral agents [4]. Naturally occurring mutations in the context of various genotypes have been identified in the structural and non-structural genes as well as regulatory elements of the virus. The best characterized mutants are the pre-core (pre-C) stop codon mutations resulting in a loss of hepatitis B e antigen [5] defined clusters of mutations in the core promoter resulting in enhanced viral replication, and mutations in the reverse transcriptase/polymerase genes conferring resistance to antivirals [6]. Furthermore, several mutations in the HBV surface gene have been identified which alter the antigenicity of the viral surface proteins (HBsAg) and structure of the viral envelope [7].
Viral hepatitis is a necroinflammatory liver disease of variable severity. Persistent infection by HBV is often associated with chronic liver disease that can lead to the development of cirrhosis and hepatocellular carcinoma (HCC) [1]. Several studies suggest that HBV is not directly cytopathic for the infected hepatocyte [1]. For example, during the early phase of HBV infection in chimpanzees (i.e., before virus-specific T cells enter the liver), 100% of the hepatocytes may be infected without histological or biochemical evidence of liver disease. Furthermore, HBV can replicate at high levels in the liver of patients when cellular immune responses are deficient or pharmacologically suppressed [8] in the absence of cytological abnormalities or inflammation [9].
2.0 Structure of The HBV Particle
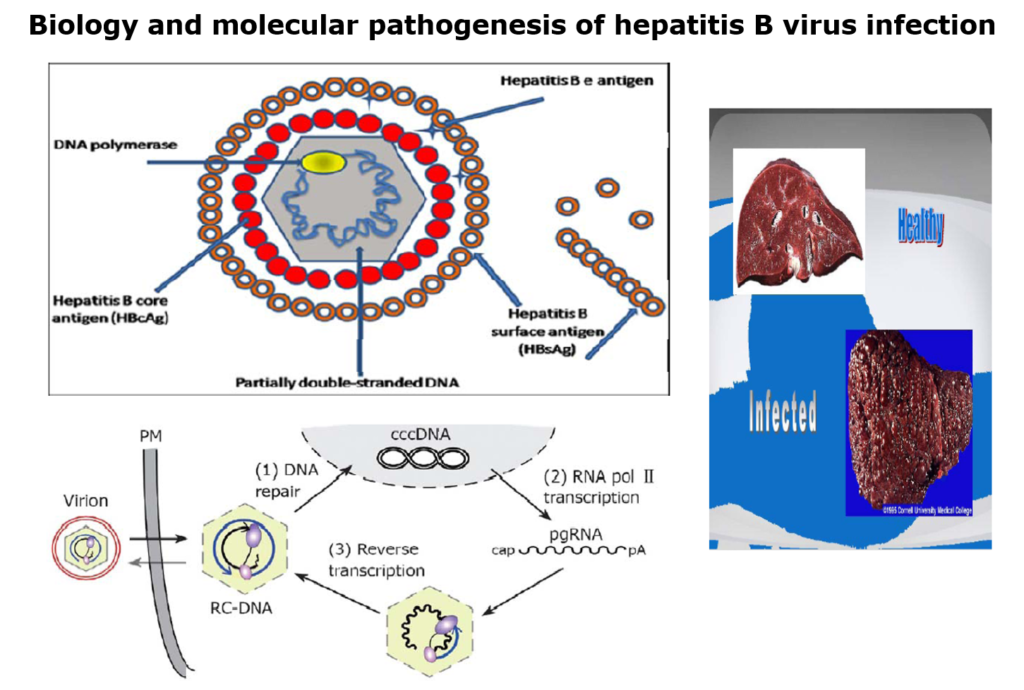
Hepatitis B virus (HBV) is a member of the Hepadnavirus family [10]. The virus particle or virion consists of an outer lipid envelope and an icosahedral nucleocapsid core, composed of protein [11]. The nucleocapsid encloses the viral DNA and a DNA polymerase that has reverse transcriptase activity. The outer envelope contains embedded proteins or the ligand which are involved in viral binding to, and entry into, susceptible hepatocytes. The virus is one of the smallest enveloped animal viruses with a virion diameter of 42 nm, but pleomorphic forms exist, including filamentous and spherical bodies, lacking a core [12]. These pleomorphic forms are not infectious and are composed of the lipid and protein that forms part of the surface of the virion, which is called the surface antigen (HBsAg), and is produced in excess during the life cycle of the virus (Figure 1).
Sources: Dane et al. [13].
3.0 Overview of the Hepadnaviral genome replication cycle
Replication of the hepadnaviral genome can broadly be divided into three phases
(1) Infectious virions contain in their inner icosahedral core the genome as a partially double-stranded, circular but not covalently closed DNA of about 3.2 kb in length (relaxed circular, or RCDNA);
(2) upon infection, the RC-DNA is converted, inside the host cell nucleus, into a plasmid-like covalently closed circular DNA (cccDNA);
(3) from the cccDNA, several genomic and subgenomic RNAs are transcribed by cellular RNA polymerase Ⅱ; of these, the pregenomic RNA (pgRNA) is selectively packaged into progeny capsids and is reverse transcribed by the co-packaged P protein into new RC-DNA genomes [14]. Matured RC-DNA containing-but not immature RNA containing-nucleocapsids can be used for intracellular cccDNA amplification, or be enveloped and released from the cell as progeny virions (Figure 2). Below we discuss these genome conversions, with emphasis on the reverse transcription step, and particularly its unique initiation mechanism [15].
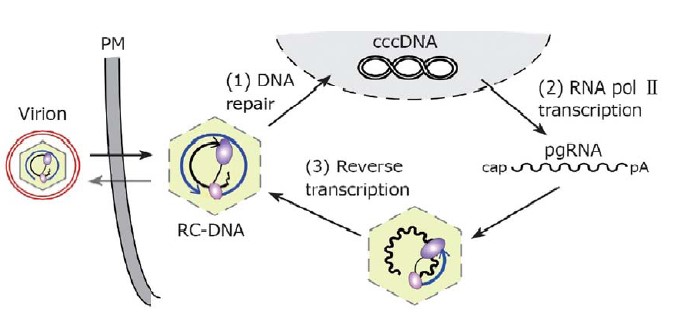
Sources: [15].
4.0 Transmission
The HBI can be transmitted by the same modes as with the human immunodeficiency virus (HIV), even though the HBV is hardier and 50-100 times more infectious than the HIV [16]. Unlike HIV, the virus can survive outside the body for at least 7 days. During that time, the virus can still cause infection if it enters into the body of a person who is not infected. Transmission of hepatitis B virus results from exposure to infectious blood or body fluids. Possible modes of transmission include but are not limited to unprotected sexual blood transfusions, re-use of contaminated needles and syringes, and vertical transmission from mother to child during childbirth. Without intervention, a mother who is positive for HBsAg confers a 20% risk of passing the infection to her offspring at the time of birth [16].
This risk is as high as 90% if the mother is also positive for HBeAg. The HBV infection can be transmitted between family members within households, possibly by contact of non-intact skin or mucous membrane with secretions or saliva containing HBV. However, at least 30% of reported hepatitis B among adults cannot be associated with an identifiable risk factor. In many developed countries (e.g. those in Western Europe and North America), patterns of transmission are different from those mentioned above. Today, the majority of infections in these countries are transmitted during young adulthood by sexual activity and injecting drug use. HBV is a major infectious occupational hazard of health workers [16].
HBV is not spread by contaminated food or water, and cannot be spread casually in the workplace. The virus incubation period is 90 days on average, but can vary from about 30 to 180 days [17]. HBV may be detected 30 to 60 days after infection and persist for widely variable periods of time.
5.0 Stages of HBV Infection
The spectrum of clinical manifestations of HBV infection varies in both acute and chronic disease. During the acute phase, manifestations range from subclinical hepatitis to anicteric hepatitis, icteric hepatitis, and fulminant hepatitis; during the chronic phase, manifestations range from an asymptomatic carrier state to chronic hepatitis, cirrhosis, and hepatocellular carcinoma. The clinical outcome of HBV infection depends upon the age at infection, the level of HBV replication, and the immune status of the host [17].
5.1 Acute HBV infection
Acute HBV infection is initial stage of the infection and every HBV- infected patient goes through this, even though not all patients transit beyond this stage [17]. Early phases of this stage of the infection are characterized serologically by the presence of HBsAg, high serum HBV DNA, HBeAg, and normal level of serum aminotransferase level (ALT), and minimal or insignificant inflammation on liver biopsy [18].
A later phase, also called immunity phase, is marked by increased serum titres of anti-HBsAg IgG (HBsAb), anti-HBcAg IgG, lowered or disappearance of HBsAg and HBV DNA, normal liver histology. This is true for those who recover fully from the infection after attaining full and permanent immunity through exposure. The duration of either phase differs among patients but generally lasts between 5 – 8 months [17]. However, those patients who fail to mobilize adequate immune response factors to combat the infection end up with the fate of living with the disease their entire lifetime. In this case, it is said the disease has become chronic. The physical signs and symptoms, such as jaundice, fever, dark-urine formation, nausea, among others, would occur, even though they will last shortly after which they get resolved following recovery. Generally, transition from the acute stage to the chronic stage depends on several factors including: age, gender, viral genotype, and host immune competence [18].
5.2 Chronic HBV infection
Chronic HBV – infected patients fall into one of the two pathologically progressive forms, namely:
a) chronic asymptomatic, marked by :
– presence of HBsAg in serum
– anti-HBsAg IgG (HBsAb ) positive
– normal liver histology indicated by apparently normal ALT levels
– relatively lower viral load (<103 copies/ml)
b) chronic symptomatic, marked by;
– presence of HBsAg in serum
– anti-HBsAg IgG (HBsAb ) positive
– relatively higher viral load (>103 copies/ml),
– significant damage on liver histology, showed by elevated ALT levels.
Patients at the chronic symptomatic stage may show mild to severe liver cirrhosis. Terminal stage of liver damage is accompanied by complications such as hepatomegaly, lower abdominal bleeding, elevated serum endogenous mercaptans, hepatic encephalopathy, hepatic coma, membranous glomerulonephritis, etc. [19].
Some studies showed that the mean annual rate of spontaneous HBeAg seroconversion ranges from 8% – 15% in children or adults with an elevated ALT level [20]. Although the ALT level is normal in most Asian children, their spontaneous HBeAg seroconversion rate is less than 2% during the first 3 years of age and then increases to 4%-5%. In some cases, spontaneous recurrence of hepatitis is not frequently recognized because it is usually asymptomatic. Since subsequent HBeAg seroconversion would not occur in such situations of hepatitis, it can thus be viewed as an abortive attempt at seroconversion [19]. However, regression of fibrosis occurs several months or years after HBeAg seroconversion [20]. The recurrence of hepatitis may precede the disappearance of HBeAg and development of HBeAg antibody, culminating in the remission of hepatitis activity [20].
5.3 Occult HBV infection
Recent investigations into the serologic and pathological distinctions among the various stages of HBV infections have led to the discovery of a new form, though rare, called occult HBV infection (HBI). Occult HBI is defined as the existence of HBV DNA in serum marked with absence of HBsAg [21]. In addition to a symptomatic and serologically evident infection, occult persistent HBV carriage has been identified since nucleic acid amplification assay enhances its sensitivity to hepadnaviral genomes and their replicative intermediates. There is evidence that occult HBV infection is a common and long-term consequence of acute hepatitis B resolution. This form of residual infection is termed as secondary occult infection (SOI) [22].
6.0 Signs and Symptoms
Acute infection with hepatitis B virus is associated with acute viral hepatitis – an illness that begins with general ill-health, loss of appetite, nausea, vomiting, body aches, mild fever, dark urine, and then progresses to development of jaundice. It has been noted that itchy skin has been an indication as a possible symptom of all hepatitis virus types. These signs may last for a few weeks and then gradually improve in most affected people. A few patients may have more severe liver disease (fulminant hepatic failure), and may die as a result of it. The infection may be entirely asymptomatic and may go unrecognized [23].
Chronic infection with Hepatitis B virus may be either asymptomatic or may be associated with cirrhosis of the liver over several years leading to accumulation of waste materials and fluids in the liver. This results in enlargement of the liver (hepatomegaly). This type of infection dramatically increases the incidence of hepatocellular carcinoma (liver cancer). Chronic carriers are encouraged to avoid consuming alcohol as it increases their risk for cirrhosis and liver cancer.

Source: [24].
7.0 HBV Diagnosis
The tests, or assays, for detection of hepatitis B virus infection involve serum or blood tests that detect either viral antigens (proteins) or antibodies produced by the host. Interpretation of these assays is complex [25]. The first step in identifying patients with chronic HBV infection is to screen those with risk factors. Screening is focused on patients in high-risk groups, such as persons born in endemic areas, patients engaged in high-risk sexual behaviors, injection drug users, dialysis patients, HIV-infected and other immunosuppressed patients, pregnant women, and persons with occupational exposure, as well as family/household members and sexual contacts of HBV-infected persons. The hepatitis B surface antigen (HBsAg) is most frequently used to screen for the presence of this infection.
Testing for antibody to hepatitis B core (anti-HBc), and antibody to hepatitis B surface antigen (anti-HBs) indicate whether an individual has been previously exposed to HBV. The HBV DNA levels are not required for preliminary screening for the HBV-infection. The HBsAg is the first detectable viral antigen to appear during infection. However, the length of time within which detectable amount of HBsAg may persist in host depends on efficiency of host immune function at clearing the virus-infected hepatocytes and establishing enduring immunity [23]. This antigen may persist when infection has become chronic stage of the infection. The molecular mechanism underlying this adaptation remains yet unknown. The infectious virion contains an inner “core particle” enclosing viral genome. The icosahedral core particle is made of 180 or 240 copies of core protein, alternatively known as hepatitis B core antigen, or HBcAg. During this ‘window’ in which the host remains infected but is successfully clearing the virus, IgM antibodies to the hepatitis B core antigen (anti-HBc IgM) may be the only serological evidence of disease.
Individuals who remain HBsAg positive for at least six months are considered to be hepatitis B carriers [19]. Carriers of the virus may have chronic hepatitis B, which would be reflected by elevated serum alanine aminotransferase levels and inflammation of the liver (Figure 3), as revealed by biopsy. Carriers who have seroconverted to HBeAg negative status, particularly those who acquired the infection as adults, have very little viral multiplication and hence may be at little risk of long-term complications or of transmitting infection to others [26]. Additionally, polymerase chain reaction (PCR) tests have been developed to detect and measure the amount of viral nucleic acid in clinical specimens. These tests measure viral loads and are used to assess a person’s infection status and to monitor treatment [27].
8.0 Pathogenesis, Virus and Host Interaction
Viral hepatitis, characterized by diffused inflammatory reaction, is associated with cell damage and death. It has been recently reported that HBV replication is associated with cell death, which is different from the widely accepted non-cytopathic characteristics of HBV [28]. The mechanism of cell damage is generally defined as the result of cytotoxic T-lymphocyte (CTL)-mediated immune responses to viral infection. Indeed, whatever the undesirable pathological effect that may result from the infection, it is only the consequence of the interaction of the virus with host factors. Generally, the hepatitis B virus interacts with the host hepatocytes at two levels [29]:
a) Virus protein interacting with host proteins (mainly transcriptional / signaling factors). b) Virus genome interacting with host proteins (mainly transcriptional factors).
9.0 Conclusions
Hepatitis B virus (HBV) is still a worldwide health concern. Hepatitis B virus (HBV) is a hepatotropic virus with the potential to cause a persistent infection, ultimately leading to cirrhosis and hepatocellular carcinoma. Over the past four decades, the basic principles of HBV gene expression and replication as well as the viral and host determinants governing infection outcome have been largely uncovered. While divergent factors are involved in its pathogenesis, it is now clear that HBV RNAs, principally templates for viral proteins and viral DNAs, have diverse biological functions involved in HBV pathogenesis. These functions include viral replication, hepatic fibrosis and hepatocarcinogenesis.
Conflict of interest: The authors declare no conflict of interest.
Authors Contributions: This work was conducted in collaboration of all authors. All authors read and approved the final version of the manuscript.
Funding: This study received no external funding
References
- Chisari, F. V., Isogawa, M., & Wieland, S. F. (2010). Pathogenesis of hepatitis B virus infection. Pathologie-biologie, 58(4), 258–266. https://doi.org/10.1016/j.patbio.2009.11.001
- Ott, J.J., Stevens, G.A., Groeger, J. and Wiersma, S.T. (2012) Global Epidemiology of Hepatitis B Virus Infection: New Estimates of Age-Specific HBsAg Seroprevalence and Endemicity. Vaccine, 30, 2212-2219.
- Walana, W., Hokey, P. and Ahiaba, S. (2014) Sero-Prevalence of Hepatitis B Virus Infection among Blood Donors: A Retrospective Study in the Kintampo Municipal Hospital, Ghana. Open Journal of Medical Microbiology, 4, 64-69.
- Baumert TF, Blum HE. Hepatitis B virus mutations: molecular biology and clinical relevance. Vir Hep Rev. 2000;6:177–192.
- Liang TJ, Hasegawa K, Rimon N, Wands JR, Ben-Porath E. A hepatitis B virus mutant associated with an epidemic of fulminant hepatitis. N Engl J Med. 1991;324:1705–1709.
- Schildgen O, Sirma H, Funk A, Olotu C, Wend UC, Hartmann H, Helm M, Rockstroh JK, Willems WR, Will H, et al. Variant of hepatitis B virus with primary resistance to adefovir. N Engl J Med. 2006;354:1807–1812
- Baumert, T. F., Thimme, R., & von Weizsäcker, F. (2007). Pathogenesis of hepatitis B virus infection. World journal of gastroenterology, 13(1), 82–90. https://doi.org/10.3748/wjg.v13.i1.82
- Guidotti LG, Matzke B, Schaller H, Chisari FV. High-level hepatitis B virus replication in transgenic mice. J Virol. 1995;69:6158–69.
- Ganem D, Prince AM. Hepatitis B virus infection–natural history and clinical consequences. N Engl J Med. 2004;350:1118–29.
- Zuckerman, A.J. (1996). Hepatitis Viruses. In: Baron’s Medical Microbiology (4th ed.). Univ of Texas Medical Branch.
- Locarnini, S. (2004). Molecular virology of hepatitis B virus.Seminars in liver disease;24 Suppl 1: 3–10.
- Howard, C.R. (1986). The Biology of Hepadna viruses. Journal of General Virology.67: 1215
- Dane, D.S, Cameron, C.G, and Briggs, M. (1970). Virus-like particles in serum of patients with Australiaantigen- associated hepatitis. Lancet 1:696-98.
- Amira, M.S.(2011)Prevalence of Hepatitis B Virus DNA Among Blood Donors in Nablus- West Bank. A Thesis is Submitted to An-Najah National University, Nablus, Palestine.
- Juergen, B. and Michael, N.(2007) Hepatitis B virus replication. World Journal of Gastroenterology. 2007; 13(1): 48-64.
- WHO (2009) Hepatitis B Vaccines. Weekly Epidemiological Record.
- Kim GA, Lim YS, An J, Lee D, Shim JH, Kim KM, Lee HC, Chung YH, Lee YS, Suh DJ. HBsAg seroclearance after nucleoside analogue therapy in patients with chronic hepatitis B: clinical outcomes and durability. Gut. 2014;63:1325–1332.
- Altiparmak, E., Koklu, S., Yalinkilic, M., Yuksel, O., Cicek, B. (2005). Viral and host causes of fatty liver in chronic hepatitis B. World journal of Gastroenterology 11: 3056–3059
- Lok, A.S., McMahon, B. (2007). Chronic hepatitis B. Hepatology (Baltimore, Md.); 45 (2):507–539.
- McMahon, B.J. (2005). Epidemiology and natural history of hepatitis B. Seminars in Liver Disease. 25 Suppl 1: 3-8.
- Torbenson, M., Thomas, D.L. (2002). Occult hepatitis B. Lancet Infect Dis; 2: 479-486
- Conjeevaram, H.S., Lok, A.S. (2001). Occult hepatitis B virus infection: a hidden menace? Hepatology; 34: 204-206.
- Lai, K., Li, N., Lui, A., Tam, T. L. (1991). Membranous nephropathy related to hepatitis Bvirus in adults. The New England journal of medicine; 324 (21): 1457–1463.
- CDC (2005). A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP).Part 2: Immunization of adults.MMWR. In press.
- Bonino, F.C., Maran P. (1987). Serological markers of HBV infectivity.Annali dell’Istituto superiore di sanita; 24 (2): 217–223
- Chu, C. and Liaw, Y. (2007). “Predictive factors for reactivation of hepatitis B following hepatitis B e antigen seroconversion in chronic hepatitis B”. Gastroenterology;133 (5): 1458–1465.
- Zoulim, F. (2006). New nucleic acid diagnostic tests in viral hepatitis.Seminars in liver disease; 26 (4): 309–317.
- Philip, M., Libbrecht, L.,Wieland, S., De Vos, R.,Habib, N., Kramvis, A.,Roskams, T., and Leroux-Roels, G. (2006). Immune Suppression Uncovers Endogenous Cytopathic Effects of the Hepatitis B Virus. Journal of . Virology. 80 (6): 2792 -2807.
- Kim, K.H., Shin, H.J., Kim, K. (2007). Hepatitis B virus X protein induces hepatic steatosis via transcriptional activation of SREBP1 and PPAR- gamma. Gastroenterology:132: 1955– 1967.













